SEMal
Implementation of "SEMal Accurate protein malonylation site predictor using structural and evolutionary information"
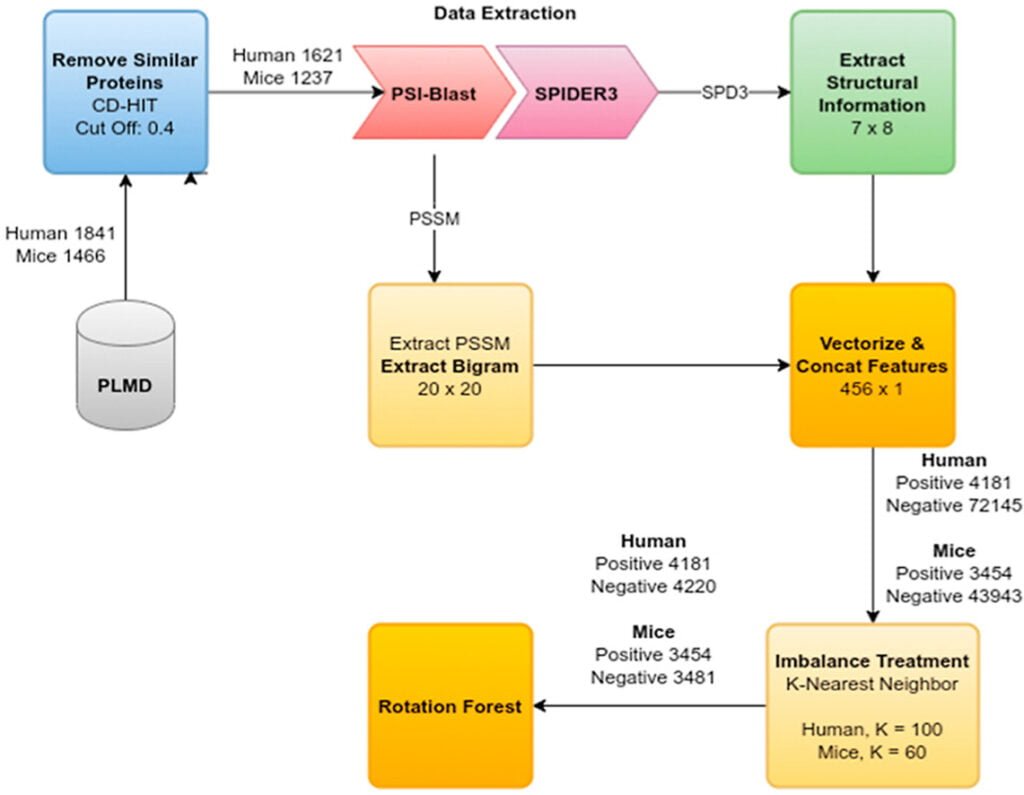
The project introduces SEMal, a novel computational approach for identifying Malonylation sites in protein sequences, vital for various biological pathways. SEMal utilizes structural and evolutionary features and employs Rotation Forest as a classifier. It outperforms previous methods in sensitivity, accuracy, and Matthews correlation coefficient for both Homo Sapiens and Mus Musculus species, offering a faster and more efficient alternative to experimental detection using mass spectrometry.
Skills: Machine Learning · TensorFlow · Scikit-Learn · Deep Learning
Published in: Computers in biology and medicine Paper Link: https://www.sciencedirect.com/science/article/pii/S001048252030353X GitHub Link: https://github.com/dipta007/SEMal